

对于二代测序的数据的处理的问题,虽然我们还是建议专业的事情。但是至少还是要了解一下基本的原理和测序当中产生的各种文件格式。由于是测序数据,其基本的原理还是基于核酸序列来进行操作的。在结果文件当中就包括两个和核酸序列位置有关的文件:VCF文件以及bed文件。
VCF和bed文件
关于这两个文件,要是想要了解怎么产生的可以去网上搜一下具体的分析流程。这里只是简单的介绍一下这两个文件都包括什么内容。
对于VCF文件主要是用来储存在测序当中发现的各种变异的情况的。比如说在测序当中发现的基因的突变或者SNP这类的。其基本的格式一共包括: 染色体号、变异发生的位置、具体的ID号、参考基因组的序列、改变后的序列。其中具体的ID这里,有的话就是ID号,没有的话可以用.来进行表示
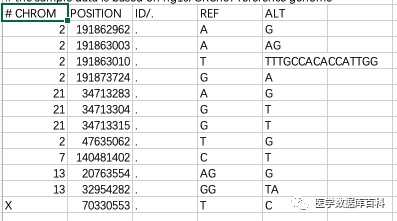
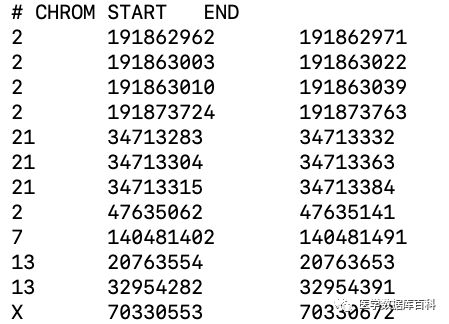
而对于bed文件而言,主要还是储存的是序列在基因组的具体位置。这类的文件主要包括三列:染色体号、序列的起始位置以及终止位置。
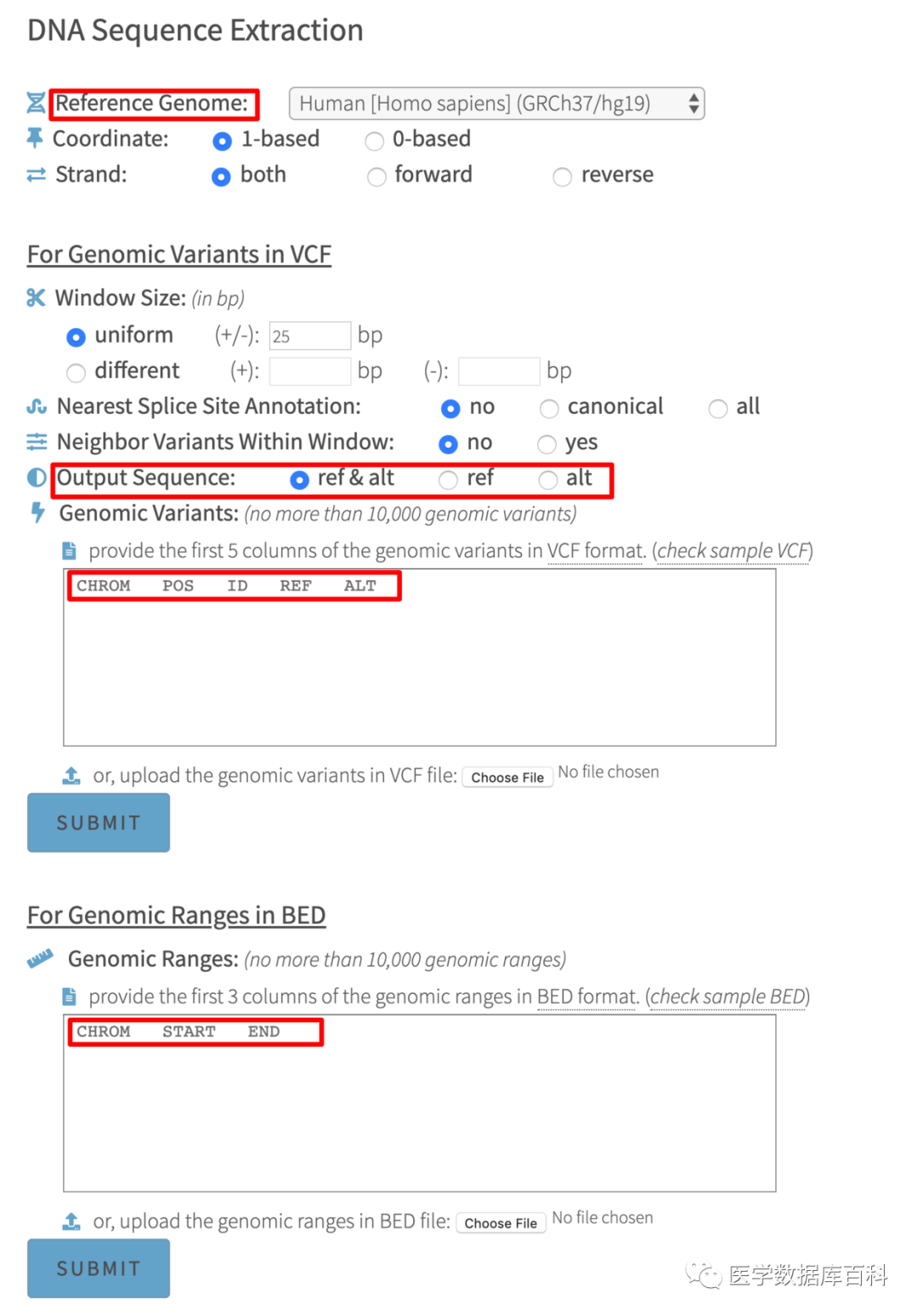
有时候我们在获得这类文件的时候,想要提取某一段位置的序列。这个时候如果好多都是基于代码行的工具来进行操作的。但是有不会代码行的工具怎么办,所以今天就给大家推荐一个在线的提取VCF/bed文件的工具。[]。这个网站可以让我们在输入VCF/BED文件之后,提取里面的具体序列。

输入
包括两个功能:提取RNA序列以及提取蛋白序列。其中提取RNA序列可以输入vcf/bed文件。而提取蛋白序列则只能输入vcf文件。

我们就以提取RNA序列来进行演示。我需要选择的是参考基因组。对于VCF数据结果,我们需要输入CHROM; POS; ID; REF; ALT。而对于bed文件。我们需要输入的则是CHROM; START; END。
另外值得注意的是,这个数据库可以预测VCF格式当中的突变对于剪切位点的关系。
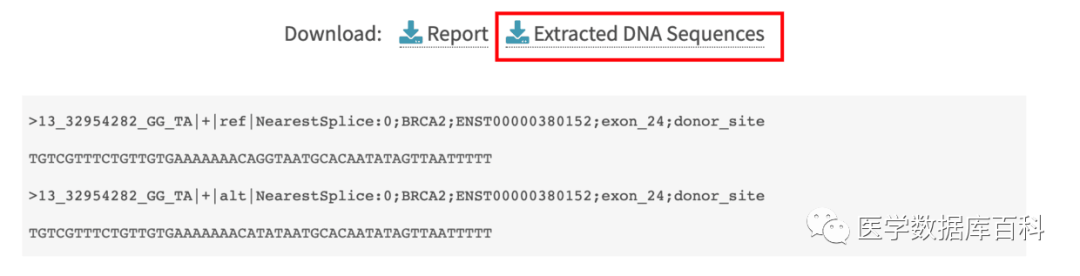
输出
这里我们输入chr13 . GG TA。同时观察这个位点的突变和剪切位点的关系。
对于剪切位点的注释:
+ve : 说明突变位于剪切位点的上游多少bp。
-ve :说明突变位于剪切位点的下游多少bp.
0: 正好位于剪切位点上。
如图则是代表正好位于剪切位点上。
工具另外使用场景
其实这个网站虽然说的是,提取测序当中VCF/BED文件的序列的。但是我们完全也可以自己来DIY的。比如我想要获得某一段基因位置的序列,那完全可以自己按照这个BED文件的格式在每一列输入相对应的内容就可以了。
———END———
限 时 特 惠: 本站每日持续更新海量各大内部创业教程,永久会员只需109元,全站资源免费下载 点击查看详情
站 长 微 信: nanadh666

